
Аномалия стебля тела плода
Обновлено: 05.10.2024
Гастрошизис представляет собой эвентрацию органов брюшной полости (чаще кишечника) через параумбиликальный дефект передней брюшной стенки. Дефект обычно располагается справа от пупка, грыжевые органы не имеют мембраны. Гастрошизис встречается с частотой 0,94 случая на 10 000 новорожденных. Частота порока у беременных моложе 20 лет выше и составляет 7 случаев на 10 000 новорожденных.
Пренатальная ультразвуковая диагностика гастрошизиса обычно основывается на визуализации петель кишечника в амниотической жидкости вблизи передней брюшной стенки плода. В некоторых случаях помимо петель кишечника за пределами брюшной полости могут находиться и другие органы. Е. Guzman одним из первых в 1990 г. сообщил о ранней пренатальной диагностике гастрошизиса с помощью трансвагинальной эхографии в 12 нед 4 дня. Выявление гастрошизисадо 12 нед беременности должно осуществляться с осторожностью, так как возможна ложноположительная диагностика в связи с наличием у плода физиологической кишечной грыжи. В эти сроки беременности убедительным доказательством гастрошизиса является четкая визуализация эвентрированных петель кишечника при условии интактного прикрепления пуповины к передней брюшной стенке плода.
В отечественной периодике, несмотря на менее частую встречаемость гастрошизиса по сравнению с омфалоцеле, к настоящему времени уже опубликовано несколько случаев пренатальной диагностики этого порока в ранние сроки беременности. По мнению М.А. Эсетова, основанием для постановки пренатального диагноза гастрошизиса в ранние сроки беременности являются: 1) обнаружение объемного образования, примыкающего к передней брюшной стенке; 2) отсутствие ограничивающей мембраны у грыжевых органов; 3) отдельный ход сосудов пуповины, не связанной с грыжевыми органами; 4) неизмененная область соединения пуповины с передней брюшной стенкой.
Согласно результатам, полученным В.Н. Демидовым, к основным эхографическим признакам, позволяющим отличить гастрошизис от омфалоцеле в ранние сроки беременности, следует отнести неправильную форму и неровный контур образования при первом из них и круглую форму, четкий и ровный контур при втором.

Аномалия развития стебля тела
Этот порок является летальным и характеризуется обширным дефектом передней брюшной стенки, выраженным кифосколиозом и рудиментарной пуповиной. Частота встречаемости аномалии составляет в среднем 1 случай на 14 000 новорожденных. Выраженные анатомические изменения при аномалии развития стебля тела позволяют диагностировать ее в конце I триместра беременности. Особо показательны в этом отношении результаты мультицентрового скринингового ультразвукового обследования 106 727 плодов в 10-14 нед беременности, проведенного в Великобритании. В ходе этого исследования было установлено, что все 14 случаев аномалии развития стебля тела были выявлены в ранние сроки беременности. Во всех диагностированных случаях верхняя половина туловища располагалась в амниотической полости, тогда как нижняя часть - в целомической полости.
Среди отечественных специалистов свои наблюдения ранней пренатальной ультразвуковой диагностики аномалии развития стебля тела опубликовали М.В. Ситарская и соавт. и Ю.Ф. Лузянин и соавт.. В наблюдении М.В. Ситарской и соавт. при ультразвуковом исследовании в 12-13 нед беременности у плода отмечены следующие изменения: позвоночник просматривался только в шейном и грудном отделах; печень, сердце и легкие находились за пределами туловища. Печеночная поверхность грыжевого образования прикреплялась к краю плаценты, вследствие чего при активных движениях плода его местоположение не изменялось.
В нашем наблюдении в ходе скринингового ультразвукового исследования в 12 нед 3 дня при трансабдоминальном доступе был выявлен обширный дефект передней брюшной стенки плода с эвентрацией большинства органов брюшной полости. При трансвагинальном ультразвуковом исследовании была достигнута более четкая визуализация структур плода и обнаружено, что позвоночник деформирован (выраженный кифосколиоз) и сформирован только в шейном и грудном отделах. Положениетуловища плода оставалось фиксированным на протяжении всего исследования, что позволило предположить наличие короткой пуповины. Дополнительное применение цветового допплеровского картирования дало возможность идентифицировать рудиментарную пуповину.
Оценка желудка плода в ранние сроки беременности необходима для исключения редких форм манифестации диафрагмальной грыжи (аномальное расположение) и обструктивных поражений верхних отделов желудочно-кишечного тракта (дилатация), среди которых нам удалось найти только два описания их пренатальной диагностики в ранние сроки беременности. G. Tsukerman и соавт. приводят описание случая пренатальной диагностики атрезии пищевода и двенадцатиперстной кишки у плода в 12 нед беременности. В ходе ультразвукового исследования плода была обнаружена выраженная дилатация желудка и двенадцатиперстной кишки. В аналогичном наблюдении Н. Blaas выявил выраженную дилатацию желудка и кишки в 10 нед бдней. После исчезновения физиологической кишечной грыжи отмечено спонтанное исчезновение дилатации кишки, желудок стал пустым, что сохранялось на протяжении всей беременности. Высказанное подозрение на атрезию пищевода подтвердилось после рождения ребенка.
В отечественной периодике впервые о преходящем образовании брюшной полости плода в ранние сроки беременности сообщила Е.А. Шевченко. В ее наблюдении первое ультразвуковое исследование было проведено в 15 нед, в ходе которого было обнаружено аваскулярное анэхогенное образование в проекции печени плода размером 19,6x8,1x3,7 мм. В 21 нед эхографическая картина образования изменилась: размеры его уменьшились, вокруг образования визуализировался венчик сниженной эхогенности, структура имела солидное строение с кальцинатами. При динамическом наблюдении в 26 нед размер образования уменьшился до 8,3x6,7x6,5 мм, структура стала более однородной и приобрела повышенную эхогенность. В 31 нед на месте образования визуализировались кальцинаты. После родов каких-либо отклонений у ребенка не отмечено. В 3,5 месяца жизни ребенку проведено ультразвуковое исследование брюшной полости, в ходе которого обнаружен кальцинат размером 4,3x3,6x1,9 мм. В 9 месяцев жизни какие-либо клинические проявления отсутствовали, психомоторное развитие соответствовало возрасту.
Мы также встретились с подобным феноменом. При трансабдоминальном ультразвуковом исследовании в 14 нед беременности в левой половине брюшной полости плода было обнаружено округлое анэхогенное образование размером 25x20x24 мм. При использовании цветового доп-плеровского картирования был исключен сосудистый генез образования. В плане дифференциальной диагностики были четко идентифицированы интактные мочевой пузырь и почки, а при применении трансвагинальной эхографии - желудок плода. Генез образования остался неустановленным. С целью комплексной пренатальной диагностики проведена трансабдоминальная аспирация ворсин хориона. Кариотип плода - 46,XY. Учитывая нормальный ка-риотип плода было решено осуществлять динамическое наблюдение, при котором в 20 и 25 нед беременности констатировано полное спонтанное исчезновение образования.
Представленные клинические наблюдения убедительно доказывают необходимость и абсолютную обоснованность выжидательной тактики в случаях обнаружения анэхогенных образований брюшной полости плода неустановленного генеза в ранние сроки беременности.
Наряду сэтим в некоторых случаях уже в 13-14 нед беременности подобными, но не преходящего характера образованиями могут быть представлены кисты печени. Так, С. Berg и соавт. приводят наблюдение пренатальной диагностики кисты печени в 13 нед. Киста визуализировалась в виде изолированного анэхогенного однокамерного образования в правом верхнем квадранте брюшной полости, ее размер составил 11x7x7 мм. В 22 нед были проведены пренатальное кариотипирование и аспирация кисты. Было удалено 18 мл темно-зеленой жидкости. Высокая концентрация желчных кислот в аспирированной жидкости и визуализация интактного желчного пузыря позволили сделать заключение о кисте печени. К моменту родов размер кисты составил 75x44x46 мм. Ребенокуспешно прооперирован на 14-е сутки жизни. При гистологическом исследовании был подтвержден пренатальный диагноз кисты печени.
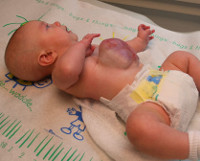
Омфалоцеле – врожденная аномалия передней брюшной стенки, при которой органы брюшной полости выходят за ее пределы в составе грыжевого мешка. Клинически патология проявляется эвентрацией петель кишечника, желудка, печени и других органов, прикрытых висцеральной брюшиной, через грыжевые ворота в участке пупочного кольца. Антенатальная диагностика включает в себя УЗИ ОБП, определение уровня α-фетопротеина, амниоцентез с дальнейшим кариотипированием. Лечение хирургическое, предусматривает радикальное или поэтапное погружение содержимого грыжевого пешка обратно в брюшную полость с последующей пластикой передней брюшной стенки.
Общие сведения
Омфалоцеле (эмбриональная грыжа, грыжа пупочного канатика) – аномалия развития, при которой происходит выпячивание органов брюшной полости, укрытых висцеральной брюшиной, сквозь переднюю брюшную стенку. Впервые данную патологию описал французский хирург А. Паре в 1634 году. В среднем заболеваемость составляет порядка 2,2:10000 новорожденных. Точную частоту, с которой встречается омфалоцеле, установить невозможно, поскольку в большинстве случаев такие беременности прерываются. У мальчиков данная патология возникает в 1,5 раза чаще, чем у девочек. Наибольшая склонность наблюдается у представителей европеоидной расы – 85% от всех случаев омфалоцеле. Для представителей негроидной расы этот показатель составляет 13%, монголоидной – 2%. Примерно 55% случаев патологии диагностируется при беременности у женщин в возрасте старше 35 лет. Общая летальность зависит от сопутствующих расстройств и колеблется от 9 до 60% новорожденных. При изолированной форме и адекватном лечении прогноз для жизни и здоровья ребенка благоприятный.
Причины омфалоцеле
Омфалоцеле – это гетерогенное заболевание, при котором нарушается процесс внутриутробного вправления физиологической пупочной грыжи. Патогенетически это может быть результатом пороков развития пупочного кольца и передней брюшной стенки, генетических аномалий, неполного погружения органов обратно в брюшную полость или дефектов строения кишечника.
При внутриутробном развитии кишечника плода, а именно – в процессе его трансформации от первичной кишечной трубки до зрелых петель происходит его физиологической разворот. Данный процесс начинается с 5 недели беременности. От этого момента и до 10 недели кишечник сильно увеличивается в объеме, из-за чего не помешается в брюшной полости. Петли давят на брюшную стенку, формируя физиологическую пупочную грыжу. К концу 10 недели абдоминальная полость стремительно прибавляет в объеме, на фоне чего происходит самостоятельное вправление эвентрации. Если данный механизм нарушается силу различных патологических изменений, возникает омфалоцеле.
Способствовать возникновению данной патологии могут вредные привычки матери (алкоголь, курение, наркотики), нерациональный прием медикаментов, беременность после 35 лет, при которой растет риск хромосомных аномалий – синдромов Патау, Эдвардса. Довольно часто омфалоцеле выступает в роли одного из симптомов таких патологий, как синдром Беквита-Видемана, пентада Кантрелла, синдром амниотических тяжей, порок развития стебля тела, OEIS комплекса. Крайне редко удается установить наследственную склонность, что свидетельствует о возможной генетической предрасположенности.
Классификация и симптомы омфалоцеле
В педиатрии в зависимости от размера дефекта и его содержания выделяют следующие формы омфалоцеле:
- Малая. Диаметр – до 5 см. Наиболее распространенная форма. Содержит 1-2 кишечные петли. В основном выступает в роли проявлений хромосомных аномалий.
- Средняя. Размер дефекта пупочного кольца – от 5 до 10 см. В составе грыжевого мешка содержатся 2-4 кишечные петли.
- Большая. Дефект передней брюшной стенки составляет более 10 см. Помимо петель кишечника через него выходит часть печени, желудок и другие органы.
По наличию сопутствующих патологий выделяют:
- Изолированное омфалоцеле. Грыжа пупочного канатика – единственная развившаяся внутриутробная патология.
- Сочетанная форма. Помимо дефекта пупочного кольца у ребенка присутствуют хромосомные мутации (25-35%), пороки развития сердечно-сосудистой (15-50%) и мочеполовой систем (до 15%). Характерны грыжи пищеводного отверстия диафрагмы и дисплазии тазобедренных суставов, другие скелетные аномалии.
Омфалоцеле является врожденной патологией, которая диагностируется еще в антенатальном периоде. После родов общее состояние ребенка зависит от сопутствующих заболеваний и срока гестации. При омфалоцеле характерны преждевременные роды, масса тела ребенка 1500г и меньше. Объективные признаки визуализируются уже с момента рождения. Определяется дефект передней брюшной стенки, который локализирован по срединной линии на уровне пупочного кольца. В этом месте находится образование, представленное петлями тонкого и толстого кишечника, возможно – желудком и печенью, покрытыми висцеральными листком брюшины. В 10-20% наблюдается анте- или интранатальный разрыв грыжевого мешка. Пуповина также входит в состав омфалоцеле. Абдоминальные мышцы развиты нормально. Другие возможные пороки развития зависят от сопутствующих патологий и могут включать в себя деформации позвоночного столба и конечностей, макроглоссию, макросомию, атрезию ануса и т. д.
Диагностика омфалоцеле
В современных условиях наличие омфалоцеле определяется еще в I триместре беременности благодаря инструментальным и лабораторным методам исследования. Ведущую роль играет УЗ-диагностика. В зависимости от аппарата установить наличие данной патологии можно уже на 11-14 неделе. Основной признак – эвентрация петель кишечника, печени, желудка за пределы брюшной полости в участке крепления пуповины к брюшной стенке. Все вышедшие органы покрыты мембраной, состоящей из брюшины, вартонового студня и амниотической оболочки. Наличие данной мембраны важно для проведения дифференциальной диагностики с гастрошизисом. При внутриутробном разрыве грыжевого мешка петли кишечника прикрывает только амнион. Также на потенциальное развитие омфалоцеле может указывать размер ворот физиологической грыжи, составляющий свыше 7 мм в диаметре. УЗИ позволяет выявить другие проявления присутствующих генетических патологий. Повторные исследования проводятся каждые 2-3 недели, т. к. возможно самостоятельно закрытие дефекта в более поздние сроки.
Лабораторная диагностика омфалоцеле осуществляется при помощи биохимического исследования крови с измерением уровня α-фетопротеина. При врожденных пороках развития этот показатель будет значительно выше нормы для имеющегося срока беременности. Для подтверждения генетических аномалий показано проведение амниоцентеза с дальнейшим кариотипированием.
Лечение омфалоцеле
При антенатальной постановке диагноза омфалоцеле роды проводятся в специализированном перинатальном центре в условиях развернутой операционной. При малых и средних размерах дефекта пупочного кольца и отсутствии угрожающих жизни матери и ребенка состояний родоразрешение может осуществляться через естественные родовые пути. Во всех других ситуациях показано кесарево сечение в связи с большим риском интранатального разрыва грыжевого мешка. Дальнейшая терапевтическая тактика зависит общего состояния ребенка, размеров пупочной грыжи и сопутствующих патологий.
Консервативная терапия используется при невозможности провести хирургическое вмешательство. Как правило, она применяется при больших формах омфалоцеле и комбинации с множественными тяжелыми аномалиями развития. Лечение заключается в формировании плотной корки и рубца, что трансформирует данную патологию в массивную вентральную грыжу, которую в дальнейшем оперируют. С этой целью применяют дубящие средства (5% перманганат калия, нитрат серебра), которые наносят на вертикально зафиксированный за пуповину грыжевой мешок 2-3 раза в день. Консервативное лечение используется крайне редко, т. к. при нем имеется большая вероятность инфицирования и сепсиса, риск разрыва оболочек, массивной спаечной болезни в дальнейшем. Предпочтение отдается раннему оперативному вмешательству.
Хирургическая тактика при омфалоцеле напрямую зависит от размеров дефекта брюшной стенки, операции могут проводиться в один или несколько этапов. Начинают такое лечение на протяжении первых 1-2 дней жизни ребенка. При одномоментном вмешательстве выполняется погружение содержимого грыжевого мешка в абдоминальную полость и послойное зашивание брюшной стенки с ее пластикой и формированием пупка. Такая тактика – метод выбора, при омфалоцеле малых и средних размеров с минимальной торакоабдоминальной диспропорцией. В других случаях прибегают к поэтапному лечению. Первый этап – подшивание силиконового мешка с силопластиковым покрытием и помещением в него содержимого грыжевого мешка. По мере постепенного погружения органов в абдоминальную полость данный мешок перевязывают, уменьшая его в объеме. На 5-15 день выполняется второй этап – удаление мешка и формирование минимальной вентральной грыжи посредством ушивания дефекта. В возрасте 5-7 месяцев эта грыжа удаляется, после чего осуществляется полноценная пластика брюшной стенки.
После первого этапа или одноэтапной операции ребенок помещается в кувез для поддержания температурного режима, применяются обезболивающие, антибиотики широкого спектра действия. Режим питания парентеральный. При необходимости выполняется декомпрессия желудка через назо- или орогастральный зонд, проводится инфузионная терапия и ИВЛ. После проведенного лечения следует длительный реабилитационный период, в течение которого осуществляется коррекция всех послеоперационных осложнений.
Прогноз и профилактика омфалоцеле
В целом прогноз для детей с омфалоцеле сомнительный. Исход зависит от срока гестации, сопутствующих хромосомных аномалий и анатомических пороков развития, диаметра дефекта пупочного кольца, объема и содержания грыжевого мешка, результатов проведенного лечения. Как правило, при адекватной терапии и отсутствии сопутствующих патологий, несовместимых с жизнью, вероятность благоприятного исхода достаточно велика. При изолированной форме омфалоцеле малого размера, несмотря возможные отдаленные осложнения хирургического лечения (ГЭРБ, спаечная болезнь, пупочная грыжа), дальнейший рост и развитие ребенка не будут отличаться от возрастной нормы. На протяжении всего периода реабилитации показано диспансерное наблюдение у лечащего педиатра, хирурга и семейного врача.
Профилактика заключается в медико-генетическом консультировании семейных пар, планировании беременности, полном отказе матери от вредных привычек. С целью ранней диагностики омфалоцеле необходимо регулярно посещать женскую консультацию и проходить соответствующие обследования: УЗИ и измерение α-фетопротеина крови.
The article describes the case of abnormal development of the fruit’s stem, leading to fetal death.
Похожие темы научных работ по клинической медицине , автор научной работы — Е.В. Морозова
Ультразвуковая Пренатальная диагностика редких врожденных пороков развития при многоплодной беременности
Анализ факторов, определяющих различия перинатальных потерь при врожденных аномалиях развития в регионах Российской Федерации (по данным государственной и ведомственной статистики)
Современный взгляд на этиопатогенез маловодия и многоводия и пути их решения при многоводии инфекционной природы
Case of prenatal examinatioin cystic - adenomatous malformations lung
Мақалада жемiстiң денесiнiң сабағының дамудың ауытқуының жағдайы жазылды.
Бауыр фиброзы жешндеп Ka3ipri кезкарас
Калиаскарова К.С., Шаймарданова Г.М., Горшкова Е.С., Кузембаева К.У., Конысбекова А.А., Ержанова А. Т., Эл'бекова К.М., Шауменова Ж.З.
Цирроздыц болжамы бауырдыц кызмет жагдайына, аскынуларына, оныц этиологиясына байланысты. Декомпенсацияланган циррозда 3 жылда наукастардыц 11- 40% Tipi болады. Прогностически неблагоприятны Гипоальбуминемия 30%-дан темен болса, протромбин индекс 50%-ден темендесе болжамы нашар болады. 1ш шеменi бар наукастардыц емip CYPУ узактыгы сирек жагдайда гана 3 жылдан асады. Бауыр комасы кезiнде елiм-жiтiм 80-100%, перитонитпен аскынса- 50%. Алкогольдi цирроз кезiнде, вирусты циррозбен салыстырганда болжамы жаксырак болады. Аутокpиндi жэне паpакpиндi факторлар фибpогенездi сапалы багалай алады.
Modern Repose on Hepatic fibrogenesis
Kaliaskarova K.S., Shaimardanova G.M., Gorshkov E.S., Kuzembayeva K.U., Konysbekova A.A.,
Erzhanova A.T., Alibekova K.M., Shaumenova Zh.Z.
Prediction of cirrhosis depends on the functional state of liver complications, its etiology. In decompensated cirrhosis after 3 years still alive 11 - 40% of patients. Prognostically unfavorable hypoalbuminemia less than 30% reduction in prothrombin index below 50%. Life expectancy of patients with ascites rarely exceed 3-5 years. Mortality in the development of hepatic coma 80-100%, with complication of peritonitis - 50%. The prognosis of alcoholic cirrhosis is better than viral. Autocrine and paracrine factors able to effectively control fibrogenesis.
СЛУЧАЙ АНОМАЛИИ РАЗВИТИЯ СТЕБЛЯ ТЕЛА ПЛОДА
Е.В. Морозова Городская больница г. Лисаковск
Введение. Аномалия развития стебля плода относится к врождённым порокам развития брюшной стенки и характеризуется отсутствием пуповины и пупочного кольца. Его развитие обусловлено неполным слиянием амниона и хориона по периферии, вследствие неправильного развития цефалической, каудальной и латеральной эмбриональных закладок туловища на 3-й недели беременности. В результате не происходит образование пуповины. Амниоперитонеальная мембрана и плацентарная хорионическая пластина сливаются на широком участке, содержимое брюшной полости персистирует в экстраэмбриональный целом. Амнион и хорион сливаются только по краю плаценты [1].
Частота этого порока, согласно литературным данным, составляет один случай на 14273 родившихся и является однозначно фатальным [1].
Аномалия развития стебля тела плода встречается в основном спорадически, но при многоплодной беременности [1]. В литературе приводится описание диагностики этой патологии в сроке 11 недель [2].
В настоящей статье привожу описание случая аномалии развития стебля тела плода, диагностированного у погибшего эмбриона в сроке 15 недель.
Материалы и методы. Беременная 36 лет направлена на ультразвуковое исследование плода врачом женской консультации, в связи с плановым обследованием при постановки на учёт. Беременность вторая. Первая беременность закончилась выкидышем в раннем сроке.
Рис.1 Аномалия развития стебля Рис.2 Аномалия развития стебля
тела плода тела плода
Выводы. Аномалия развития стебля тела плода может развиваться не только в случае многоплодной беоременности и может служить причиной внутриутробной гибели плода в ранние сроки беременности. В случае выявления, при скрининговом осмотре в сроке 10-11 недель, близкого расположения плода к плаценте следует делать повторное ультразвуковое исследование до 15 недели беременности.
Жемютщ денесшщ сабагыньщ дамудьщ ауыткуыньщ жагдайы
Макалада жемютщ денесшщ сабагыньщ дамудьщ ауыткуыньщ жагдайы жазылды.
Case of prénatal examinatioin cystic - adenomatous malformations lung
The article describes the case of abnormal development of the fruit's stem, leading to fetal death.
2. Стыгар А.М., М.В. Медведев Ультразвуковое исследование плаценты, пуповины и околоплодных вод. //Клиническое руководство по ультразвуковой диагностике/ Под ред. Митькова В.В., Медведева М.В. М.: Видар 1996. Т. 2. С. 71-72.
Медико-генетическое отделение Московского областного НИИ акушерства и гинекологии, Москва.
Курс пренатальной диагностики, ФГБОУ ДПО РМАПО Минздрава России, Москва.
УЗИ сканер HS50
Доступная эффективность. Универсальный ультразвуковой сканер, компактный дизайн и инновационные возможности.
Настоящая работа посвящена изучению плодового фенотипа при ультразвуковом исследовании в различные сроки беременности при помощи новейших технологий Samsung, оптимизирующих диагностику многих врожденных пороков развития, аномалий, нарушений строения, особенностей развития с оценкой значимости отдельных ультразвуковых патологических маркеров в качестве потенциальных признаков генетических синдромов и ассоциаций.
Одной из ведущих причин детской и младенческой смертности являются врожденные пороки развития (ВПР), среди которых в 20% случаев встречаются множественные ВПР (МВПР) с установленной частотой 1:250 новорожденных [1]. Особое значение в профилактике МВПР имеет пренатальная диагностика – современный и высокоэффективный метод диспансеризации плода, направленный на своевременное выявление патологии, впоследствии определяющий выбор адекватной акушерской тактики при данной беременности и специфических мер профилактики болезни в данной семье в дальнейшем [2]. Современные ее возможности позволяют из комплекса патологических нарушений у плода выделить достоверно значимые ультразвуковые маркеры для постановки диагноза генетических синдромов различной этиологии. Сегодня пренатальная синдромология активно развивается, как в плане расширения спектра возможных лабораторных исследований, так и описания значимых, ключевых, таргетных ультразвуковых признаков многих наследственных синдромов различного происхождения.
Группа МВПР включает сотни нозологических форм, различных по этиологии, фенотипическим проявлениям и прогнозу. В силу огромной гетерогенности МВПР представляют собой сложную проблему для постановки окончательного диагноза. Основными этиологическими факторами, приводящими к возникновению синдромов с множественными пороками развития, являются генные, хромосомные мутации и действие на плод неблагоприятных факторов внешней среды [3, 4].
Любые наследственные или врожденные генетические болезни являются результатом повреждения генетической информации на геномном, хромосомном или генном уровне. В 60% всех случаев МВПР встречаются хромосомные синдромы, достаточно широко изученные и хорошо диагностируемые. Для лабораторной диагностики этих повреждений разработаны разнообразные и хорошо известные методы: цитогенетический, молекулярно-цитогенетический и молекулярно-генетический. МВПР нехромосомного генеза встречаются в 40% случаев. Среди них выделяют синдромы, ассоциации и неклассифицированные комплексы. Применительно к нехромосомным синдромам цитогенетическое исследование позволяет лишь отвергнуть хромосомную природу данного комплекса пороков развития, но не установить диагноз.
Сегодня имеется определенное количество публикаций о выявлении некоторых синдромов и ассоциаций при пренатальной эхографии [6, 7]. Однако дородовая идентификация нозологических форм нехромосомных синдромов пока не вошла в рутинную практику врачей в области пренатальной диагностики и носит случайный характер. В литературе отсутствуют четкие рекомендации о системном подходе к диагностике наследственных синдромов различной этиологии, нет диагностических алгоритмов и определенных схем взаимодействия врачей ультразвуковой пренатальной диагностики, генетиков, акушеров-гинекологов.
Революцией в пренатальной ультразвуковой диагностике явилось появление объемной эхографии, которая, обладая такими качествами, как неинвазивность, безопасность и возможность многократного применения у одной пациентки, имеет высокую информативность в исследовании анатомии плода и изучении его фенотипа. При применении различных режимов объемной эхографии абсолютно очевидно их преимущество по сравнению с обычным сканированием. Детально можно изучить лицо плода (рис. 1–4) в различные сроки беременности, начиная со сроков первого пренатального скрининга в 11–14 нед, конечности плода, причем не только их наличие и положение (рис. 5, 6), но и состояние и количество пальцев (рис. 7–9) как на руках, так и на ногах. Также можно изучить позвонки плода (рис. 10), состояние твердого нёба (рис. 11, 12), строение наружного уха (ушной раковины) (рис. 13), состояние основных швов черепа и родничков, исключая их преждевременное закрытие при кранисиностозах (рис. 14, 15).
Городская Ивано-Матренинская детская клиническая больница, Иркутск; Иркутская государственная медицинская академия последипломного образования
Городская Ивано-Матренинская детская клиническая больница, Иркутск; Иркутский государственный медицинский университет; Иркутская государственная медицинская академия последипломного образования
1. Городская Ивано-Матренинская детская клиническая больница, Иркутск; 2. Иркутский государственный медицинский университет; 3. Иркутская государственная медицинская академия последипломного образования
Городская Ивано-Матренинская детская клиническая больница, Иркутск
Врожденные дефекты брюшной стенки
Журнал: Хирургия. Журнал им. Н.И. Пирогова. 2016;(5): 74-81
Городская Ивано-Матренинская детская клиническая больница, Иркутск; Иркутская государственная медицинская академия последипломного образования



Городская Ивано-Матренинская детская клиническая больница, Иркутск; Иркутская государственная медицинская академия последипломного образования
Городская Ивано-Матренинская детская клиническая больница, Иркутск; Иркутский государственный медицинский университет; Иркутская государственная медицинская академия последипломного образования
1. Городская Ивано-Матренинская детская клиническая больница, Иркутск; 2. Иркутский государственный медицинский университет; 3. Иркутская государственная медицинская академия последипломного образования
Городская Ивано-Матренинская детская клиническая больница, Иркутск
Врожденные аномалии передней стенки брюшной полости делятся на омфалоцеле и гастрошизис. Несмотря на то что они рассматриваются всегда вместе, природа этих заболеваний различна. Основные отличия представлены в таблице [1].

Характеристика врожденных пороков развития передней брюшной стенки
Эмбриология и этиология. Брюшная стенка начинает формироваться с 4-й недели внутриутробной жизни в результате разнонаправленного роста тела эмбриона в краниокаудальном и медиолатеральном направлениях. Во время 6-й недели быстрый рост печени и кишечной трубки вызывает перемещение средней кишки в пупочный канатик. В последующие 4 нед происходит процесс удлинения и ротации средней кишки. К 10-й неделе кишка возвращается в брюшную полость, где первые три отдела двенадцатиперстной кишки, восходящая и нисходящая толстая кишка прикрепляются в задней поверхности брюшной полости и перемещаются в ретроперитонеальную позицию.
Современное понимание происхождения омфалоцеле базируется на популярной теории, объясняющей это явление как нарушение процесса возвращения органов в брюшную полость. Это предположение подтверждают находки в грыжевом мешке, состоящем из листков брюшины, вартонова студня и амниона, различных внутренних органов, таких как печень, мочевой пузырь, желудок, яичник, яички.
Гастрошизис
Пренатальный диагноз. Гастрошизис встречается у 1 из 4000 появившихся на свет новорожденных [6]. Документально подтверждено увеличение числа новорожденных с гастрошизисом у матерей моложе 21 года [7]. Повсеместно во всем мире отмечено увеличение новорожденных с этим заболеванием [8]. Недоношенность также существенно чаще встречается у больных с гастрошизисом, чем в обычной популяции детей (28% против 6%) [9].
Это заболевание оказывает очевидное и значительное влияние на пациентов, их семьи и систему здравоохранения в целом. Новорожденные с гастрошизисом нуждаются в длительной госпитализации после рождения, которая составляет от 25 до 50 дней при стоимости лечения от 70 000 до 150 000 долл. США 11. Более 95% пациентов имеют шанс на выживание благодаря улучшению терапии, которая сосредоточена на совершенствовании хирургических технологий и периоперативного лечения [10, 15, 16].
Осложнение беременности гастрошизисом у плода в большинстве наблюдений диагностируют до 20-й недели гестации. Ультразвуковое исследование проводят после того, как у беременной женщины определяется повышенный уровень альфа-фетопротеина [19, 20]. Свободно плавающие в амниотической жидкости кишечные петли и дефект брюшной стенки, расположенный справа от пуповины, являются достоверными признаками заболевания.
С помощью пренатального ультразвукового исследования стало возможным получить дородовую информацию о пороке развития передней брюшной стенки. Оценке подвергается объем амниотической жидкости (полигидрамнион связан с осложненными формами заболевания), окружность брюшной полости (прогнозирование вместимости брюшной полости) и увеличение диаметра кишки (предсказание кишечной атрезии). Большинство авторов рекомендуют преждевременное родоразрешение, если у плода обнаруживают расширенные и утолщенные петли кишечника. Однако довольно часто исследования дают ложноположительный результат. Иногда расширение кишечника является результатом временных функциональных или механических преград, исчезающих после рождения. Кроме того, диаметр расширенной тонкой кишки может иметь различные значения, и их интерпретация затруднена тем, что дилатация кишечника часто не появляется до 32-36 нед беременности.
Перинатальное лечение. Оптимальный срок родоразрешения и его способ при гастрошизисе до сих пор не определены. Приверженцы кесарева сечения мотивируют свой выбор опасностью повреждения петель кишки и повышенным риском развития инфекции и сепсиса [21], однако в большинстве источников литературы отмечено, что способ родоразрешения не влияет на исход заболевания [28, 29] и оба метода одинаково безопасны. Преждевременное родоразрешение является другим важным вопросом, который обсуждается при выявлении пациентов с гастрошизисом [30]. В экспериментах обнаружено высокое содержание интерлейкина-6, интерлейкина-8, ферритина в амниотической жидкости, в которой находятся эмбрионы с гастрошизисом [31, 32]. Считается, что длительное воздействие амниотической жидкости на кишечник приводит к повреждению интестинальных пейсмейкеров и параличу нервных сплетений, вызывающих продолжительное нарушение перистальтики и абсорбции нутриентов [33]. Ранние роды могут предупредить эти явления, однако сведения литературы на этот счет недостаточны [33]. Исследование ученых из Великобритании не подтвердило ожидаемого влияния преждевременных родов на исход гастрошизиса [34]. Более того, другая научная работа продемонстрировала, что рождение детей с массой тела менее 2 кг сопровождается повышенной летальностью [35]. Таким образом, современный уровень знаний не поддерживает преждевременное родоразрешение при наличии плода с гастрошизисом.
У пациентов с гастрошизисом при рождении имеются существенные потери жидкости, которые происходят с поверхности брюшины и петель кишечника, расположенных снаружи. Инфузия жидкости должна быть налажена сразу после рождения. В полость желудка обязательно устанавливают назогастральный зонд, предотвращающий дальнейшую дилатацию желудка и кишечника. Кишечник необходимо укрыть салфеткой, смоченной теплым физиологическим раствором, и переместить в центральное положение. Новорожденного следует размещать на правом боку для предупреждения перегиба брыжейки и ишемии кишки. Кишечник оптимально поместить в пластиковый мешок. В качестве альтернативы пациент может быть полностью помещен в пластиковый пакет для снижения потерь жидкости с поверхности органов и поддержания температурного баланса. При осмотре кишечной трубки необходимо уделить внимание поиску сопутствующих аномалий, таких как атрезия, некроз или перфорация кишки [42].
Хирургическое лечение. Главная цель лечения заключается в возврате органов в брюшную полость. Этот процесс должен сопровождаться минимальным риском повреждения кишки и повышения внутрибрюшного давления. Используют две наиболее популярные стратегии лечения гастрошизиса - первичную репозицию [43] и этапное погружение. Время и место этих процедур продолжают обсуждать [44]. Во всех наблюдениях необходимо обследование кишечника на предмет обнаружения атрезии, некроза и перфорации. В ходе первичного осмотра могут быть обнаружены спайки, которые пересекают кишечные петли, их подвергают рассечению до того, как кишечник будет погружен в брюшную полость.
Этапное лечение. Первоначально этот способ реализовали путем вшивания в края дефекта протеза, изготовленного из силастиковой пластины. В середине 90-х годов прошлого века был разработан фабричный силастиковый протез с циркулярным кольцом нескольких типовых размеров (5 и 7,5 см), которое размещали под фасцию без наложения апоневротических швов и использования общей анестезии [58]. Процедуру погружения внутренних органов стало возможным выполнять непосредственно в родильном зале или у постели больного. После установки силастикового протеза кишечник ежедневно дозированно погружали в брюшную полость, а резервуар уменьшали в размерах путем простой перевязки избытка пластикового пакета выше кишечных петель, находящихся в нем. Когда содержимое полностью перемещалось в брюшную полость, производили кожно-фасциальное закрытие дефекта. Этот процесс обычно занимает от 1 до 14 дней и обычно длится около 1 нед, все зависит от состояния кишечника и самого ребенка. Окончательное закрытие производили в операционном зале: мобилизовывали кожные и фасциальные листки, которые затем ушивали в горизонтальном или вертикальном направлении. Герметизацию кожного дефекта производили чаще всего в поперечном направлении с формированием кожного шва в виде замочной скважины - горизонтального рубца справа от пупка.
Послеоперационное лечение. Гастрошизис сопровождается нарушениями моторики кишечника и абсорбции нутриентов, которые обнаруживают почти у всех пациентов в послеоперационном периоде. Полное энтеральное питание очень часто задерживают на несколько недель в ожидании восстановления нормальной функции кишечника. В течение этого периода ожидания выполняют назогастральную декомпрессию желудка и назначают полное парентеральное питание. Когда появляется моторная активность кишечной трубки, возобновляют энтеральный прием пищи, который медленно расширяют. Очень важно при этом наличие центрального венозного доступа, так как восстановление энтерального питания может занимать несколько недель. Этим пациентам могут быть полезны препараты, усиливающие перистальтику кишечника. В эксперименте на новорожденных кроликах один из них - цизаприд - улучшал сокращение кишечника, в то время как другой - эритромицин - усиливал подвижность кишечника в контрольной группе взрослых кроликов [68]. В то же время рандомизированное исследование прокинетических эффектов эритромицина не продемонстрировало его влияние на ускорение процесса перехода пациентов на полное энтеральное питание [69].
Крипторхизм обнаруживают у 15-30% больных с гастрошизисом. Ретроспективное исследование показало, что возвращение в брюшную полость яичек чаще всего приводит к их нормальному опущению в полость мошонки [75]. Большинство исследователей сообщают, что процесс опускания яичек в мошонку происходит в течение первого года жизни. Только после выжидания этого времени стоит выполнять орхипексию [76]. Если в результате произведенных реконструкций брюшной стенки у больных не формируется пупочное кольцо, то до 60% детей сообщают о наличии у них психологического стресса, связанного с отсутствием пупка [77] .
Омфалоцеле
Перинатальное лечение. Способ родоразрешения при наличии у детей омфалоцеле должен быть продиктован акушерскими соображениями, так как прежние представления о безопасности кесарева сечения оказались неподтвержденными [87]. Беременность чаще всего продляют до полного срока и предпочитают рекомендовать самостоятельные вагинальные роды. Однако в некоторых наблюдениях при гигантском омфалоцеле рекомендовано кесарево сечение из-за опасности повреждения печени.
Лечение в родильном доме. После рождения ребенка проводят тщательный поиск сопутствующих аномалий, что является очень важным. Все новорожденные должны пройти ультразвуковое исследование грудной клетки и брюшной полости для поиска дефектов развития сердца и почек. Новорожденных необходимо обследовать на гипогликемию, чтобы исключить синдром Беквита-Видемана (Bechwith-Wiedemann). Также рекомендуется взять пробу крови для генетического исследования. С целью подготовки пациентов с омфалоцеле к транспортировке должны быть оценены все риски, связанные с сопутствующими аномалиями. Чаще всего у больных с омфалоцеле не бывает больших потерь жидкости и колебаний температуры, которые наблюдаются у больных с гастрошизисом, но все-таки они встречаются и более выражены, чем у обычных новорожденных. Невзирая на это, оболочки омфалоцеле следует укрыть влажной салфеткой для еще большей минимизации потерь жидкости. В заключение в желудок устанавливают назогастральный зонд. В некоторых наблюдениях оболочки омфалоцеле могут быть повреждены во время родов или сразу после рождения. Большое омфалоцеле с повреждением оболочек является одной из сложных хирургических проблем. В этих ситуациях цель лечения заключается в экстренном укрытии внутренних органов. Только немногие исследования демонстрируют хороший исход при этих состояниях, сопровождающихся высокой частотой развития кишечных свищей, сепсиса и легочной гипоплазии [88].
Неоперативное лечение омфалоцеле. Нехирургические техники в основном базируются на применении химических агентов, наносимых поверх грыжевого мешка и способствующих развитию коагуляционного струпа. Эпителизация дефекта происходит в течение длительного времени и приводит к формированию вентральной грыжи. Этот подход используют в настоящее время очень редко, скорее всего, в тех случаях, когда новорожденные имеют серьезные проблемы со стороны легких и сердца. Эта концепция не нова и появилась задолго до первого описания R. Gross [97] метода, при котором используют кожный лоскут для формирования вентральной грыжи.
Послеоперационное лечение. Если была предпринята первичная реконструкция при омфалоцеле, то большинство пациентов нуждаются в продленной ИВЛ. Питание может быть предложено только тогда, когда появляется моторная активность кишечника. Антибиотики назначают на период не меньше 48 ч после операции и, если существуют признаки инфекции, введение продолжают. В настоящее время вопрос о том, какой метод лечения омфалоцеле приводит к сокращению сроков госпитализации, не может быть разрешен, так как сравнивающиеся серии насчитывают ограниченное число больных [90]. Установлено, что время, необходимое для перехода на полное энтеральное питание, было короче в группе детей после первичного закрытия. В одном из обзоров лечения пациентов с омфалоцеле авторы сообщают об осложнениях у 12% больных после первичного закрытия (инфаркт кишечника, почечная недостаточность, острое переполнение печени кровью) [100].
Читайте также: