
Синдром денди уокера на узи плода описание
Обновлено: 05.10.2024
Лысова Ю.В.
Научный руководитель: к.м.н., доцент Нечаев В.Н.
Резюме
Количество врожденных пороков развития в последнее десятилетие заметно увеличилось. Пороки развития нервной системы суммарно занимают третье место в структуре аномалий развития после врожденной патологии сердечно-сосудистой и мочевыводящей систем, причем около 80% этих заболеваний представлены гидроцефалией различного генеза [1]. Среди большого количества возможных аномалий одним из наиболее тяжелых по своим последствиям считается синдром Денди Уокера.
Ключевые слова
Статья
Данный синдром был впервые описан американским нейрохирургом Уолтером Денди в 1921 году и Эрлом Уокером в 1944 г. Среди живорождённых детей частота встречаемости от 1:5000 до 1:25000, а среди детей с врождённой гидроцефалией колеблется от 3,5 до 12% [2].
Синдром Денди-Уокера (Dandy-Walker Syndrome) - это порок развития головного мозга (мозжечка и окружающих его ликворных пространств), для которого характерна триада симптомов: гипотрофия червя мозжечка и/или полушарий мозжечка, кисты задней черепной ямки, гидроцефалия различной степени. Данный синдром по МКБ Х кодируется в рубрике аномалий развития под шифром Q 03.1, как атрезия отверстий Мажанди и Люшка [3, 4].
Согласно современным представлениям этиология синдрома Денди-Уокера чрезвычайно гетерогенна, так как в его возникновении принимают участие разные факторы: наследственные (хромосомные и генные) и экзогенные тератогены; предполагается, что определенную роль играют и такие факторы как вирусная инфекция (ЦМВ, краснуха); приём алкоголя, диабет беременно. В 1/3 – 1/2 случаев синдром Денди-Уокера сочетается с другими различными врожденными синдромами (табл. 1) [2].
В 70% случаев порок сочетается и с другими аномалиями головного мозга — агенезией мозолистого тела, энцефалоцеле, полимикрогирией, агирией, гетеротопией серого вещества, а также с поражениями других органов и систем (полидактилией, синдактилией, врожденными пороками сердца, поликистозом почек, расщелинами неба и др.) [5].
Среди основных гипотез возникновения синдрома Денди-Уокера можно выделить следующие:
- остановка эмбрионального развития в процессе формирования ромбовидного мозга;
- атрезия выходного отверстия из IV желудочка при отсроченном открытии отверстия Мажанди;
- возникновение сосудистого сплетения IV желудочка в середине тонкой крыши ромбовидного мозга.
Синдром Денди-Уокера характеризуется выраженным клиническим полиморфизмом: от практически нормального постнатального развития до тяжелой инвалидности и даже гибели ребенка. Перинатальные исходы во многом зависят от глубины поражения ЦНС (прогрессирующая гидроцефалия), а также от наличия сочетанной патологии, которая в 60 - 75% случаев сопровождает данный синдром [2, 5].
Прогноз для жизни и здоровья при синдроме Денди — Уокера зависит от наличия сочетанных аномалий развития, хромосомных аномалий и срока диагностики. По данным литературы, показатели постнатальной заболеваемости и смертности выше в тех случаях, когда синдром диагностирован в пренатальном периоде, а не постнатально [2] .
Лечение данного заболевания симптоматическое. При наличии признаков нарастающей внутричерепной гипертензии проводят шунтирующие операции. Исход часто летальный, в 90% случаев это первые годы жизни [7].
Под наблюдением находился доношенный новорождённый мальчик Л., родившийся от 3 беременности, протекавшей на фоне поздней постановки на учёт (в 30 недель). С 35 недель гестации отмечалось многоводие и впервые по данным ультразвукового исследования выявлена аномалия развития плода - мальформация Денди-Уокера.
Картина пренатального УЗИ в 35 недель гестации: форма головы аномальная (клубникообразная). Кости при надавливании датчиком не деформируются. Расширение боковых желудочков, передние рога до 10 мм справа и слева. Полость прозрачной перегородки до 9,9 мм. Задние рога – 11 мм, слева – 12 мм. Отмечено расширение IVжелудочка до 12 мм. Имеется гипоплазия червя мозжечка, расширение большой цистерны до 12 мм.
Ребенок от третьих срочных родов в головном предлежании. Околоплодные воды светло-жёлтые в объеме двух литров. Масса тела ребёнка при рождении составила 2890 г, рост - 49 см., окружность головы - 35 см., груди - 33 см. Оценка по шкале Апгар 4 - 7 баллов.
Состояние ребёнка после рождения тяжелое, из родильного зала поступил в ОРИТН, за счёт дыхательной недостаточности III степени и выраженной неврологической симптоматики. C момента поступления находился на ИВЛ, с третьих суток жизни был переведен на респираторную поддержку в режиме СРАР, а с 7 суток жизни на спонтанное дыхание. Оксигенотерапия проводилась по показаниям. Энтеральное питание начато с первых суток в трофическом объеме с последующим расширением, усваивал.
Обращало на себя внимание наличие двусторонней косолапости с варусной деформацией обеих стоп, кистей и фаланг пальцев, гипертелоризма, седловидной переносицы, нависающего лба и затылка, широкого языка, короткой шеи (рис. 3).
На 14 сутки жизни, после стабилизации состояния, ребёнок был переведён на второй этап лечения, в отделение патологии новорожденных для дальнейшего наблюдения и лечения.
В динамике по данным НСГ выявлена перивентрикулярная ишемия, подтверждён синдром Денди-Уокера, вентрикуломегалия (как часть симптомокомплекса) и повышенная резистентность сосудов мозга.
Ребёнок был осмотрен неврологом, окулистом, генетиком, выставлен диагноз: Врожденный порок развития ЦНС - синдром Денди Уокера. Гипоксически-ишемическое поражение ЦНС. После осмотра нейрохирурга выставлен диагноз: ВПР ЦНС синдром Денди-Уокера, гидроцефальный синдром.
На момент осмотра в нейрохирургическом лечении не нуждался, были даны рекомендации по уходу и лечению.
По результатам ДЭХО-КГ выявлен врожденный порок сердца: комбинированный стеноз легочной артерии (клапанно-подклапанный), ДМПП со сбросом слева направо. Открытое овальное окно диаметром 2,0 мм.
Ребенок был консультирован кардиологом и кардиохирургом, даны рекомендации.
По данным УЗИ брюшной полости и почек патологии не выявлено.
После осмотра врача-ортопеда была подтверждена врожденная двусторонняя косолапость.
В связи со стабилизацией состояния ребёнка, после проведённого обследования и лечения, а также отказа матери от родительских прав, мальчик на 57 сутки жизни был переведён в дом ребёнка города Маркса.
В заключение следует сказать о том, что проведённое настоящее наблюдение представляет большой интерес с клинической точки зрения, поскольку встречается ни так часто в повседневной практике врача.
Ранняя диагностика сложных генетических синдромов, к коим относится и описываемое клиническое наблюдение, представляет определенные сложности. По моему мнению, в подобных ситуациях оправдана постановка синдромального диагноза с уточнением аномалий развития на основании анализа совокупности клинических данных, дополнительных методов обследования. Точный нозологический диагноз важен не только для генетического анализа, медико-генетического консультирования, но и, прежде всего, для профилактики и лечения. Без достоверного клинического диагноза невозможен анализ факторов и их теоретическое осмысление.
Литература
2. Барашнев Ю.И. Перинатальная неврология. – М.: Триада-Х, 2001.- С. 181-232.
4. Неонатология (национальное руководство) / Под ред. Н.И.Володина. - М., ГЭОТАР-Медиа, 2007; 847 с.
5. Наследственные болезни / Под ред. Л.О.Бадаляна. – Т.: Медицина, 2002.- С. 138.
6. Пренатальная диагностика наследственных и врожденных болезней / Под ред. Э.К.Айламазяна, В.С.Баранова. – М.: Триада-Х, 2007. – С. 11-148.
7. Барашнев Ю.И., Бахарев В.А., Новиков П.В. Диагностика и лечение врожденных и наследственных заболеваний у детей. М.: Триада-Х, 2004.- С.12-87.
Таблицы
Синдромы, с которыми сочетается гидроцефалия Денди-Уокера
Синдромы
Клинические признаки
Этиология
Различные хромосомные аномалии
Поражение различных органов и систем
Хромосомные делеции, дупликации, трисомии, триплоидии
Лиссэнцефалия, ретинальная дисплазия, аномалия глаз, энцефалоцеле, миопатии и др.
Мутации генов POMT1 (локализации 9g34.1), POMT2 (14g24.3), FKTN (9g31) и др. Тип наследования аутосомно-рецессивный
3С синдром (краниомозжечково-сердечная дисплазия, Ritscher-Schinzel синдром)
Задержка роста, пороки сердца (септальные дефекты), гипертелоризм и др.
Ген не установлен. Тип наследования аутосомно-рецессивный
Артериальные аномалии, включая коарктацию аорты, дефекты сердца, глаз (микроофтальмия).
Синдром Денди-Уокера – это врожденная патология нервной системы, для которой характерна триада признаков: гидроцефалия, гипоплазия или аплазия мозжечка, кисты задней черепной ямки. Болезнь имеет полиэтиологическую природу, среди провоцирующих факторов выделяют генетические аномалии, тератогенные влияния. Клиническая симптоматика включает классические признаки гидроцефалии, многообразные неврологические нарушения. Диагностика порока требует проведения нейросонографии, МРТ, эхокардиографии, также возможна пренатальная постановка диагноза при УЗИ-скрининге. Лечение заключается в консервативных и хирургических методах коррекции гидроцефалии.
МКБ-10
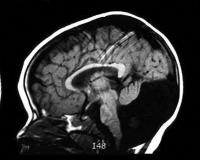


Общие сведения
Пороки ЦНС занимают второе место в группе врожденных болезней, уступая только сердечно-сосудистым аномалиям. До 80% всех патологий сопровождаются гидроцефалией, как при синдроме Денди-Уокера. Данное заболевание было описано американским нейрохирургом В. Денди в 1914 году, спустя 28 лет канадско-американский нейрохирург А. Уокер с коллегами предложил оперативный способ лечения порока. Патология встречается с частотой 1:25000-1:35000, девочки болеют чаще. Среди младенцев с врожденной гидроцефалией патология регистрируется в 3,5-12% случаев.
Причины
Этиологические факторы синдрома Денди-Уокера до сих пор точно не выяснены. По современным данным, патология имеет мультифакториальную природу, ее развитию способствуют как внутренние причины (хромосомные и генные мутации), так и воздействие экзогенных тератогенов. Провоцирующими факторами заболевания могут выступать:
- вирусные инфекции (цитомегаловирус, краснуха);
- гестационный диабет;
- употребление алкоголя матерью во время беременности.
Новейшие исследования показывают четкую связь изолированной формы аномалии Денди-Уокера с мутациями генов ZIC1 и ZIC4. Риск развития заболевания повышается у младенцев с врожденными нарушениями обмена веществ. Чаще всего синдром ассоциирован с одной из форм 3-метилглутаконовой ацидурии, которая проявляется ретинопатией, почечной дисфункцией, метаболическим ацидозом и повышением активности печеночных ферментов.
Патогенез
Выделяют три основные гипотезы структурных поражений головного мозга при болезни Денди-Уокера. Согласно первой из них, пороки ЦНС вызваны замедлением эмбрионального развития на раннем этапе при закладке ромбовидного мозга. Такая ситуация наблюдается под действием тератогенных факторов либо на фоне генетических мутаций. Вторая гипотеза указывает на заращение выходного отверстия 4 желудочка мозга и позднее открытие апертуры Мажанди.
Новые экспериментальные данные подтверждают вероятность третьей гипотезы: типичные анатомические изменения развиваются при возникновении сосудистого сплетения на тонкой крыше ромбовидного мозга. Патология сопровождается внутриутробной гидроцефалией, вызывает серьезные неврологические нарушения. На фоне этого развивается большая киста в задних отделах черепа, которая препятствует формированию мозолистого тела и червя мозжечка.
Классификация
Специалисты выделяют открытую и закрытую формы синдрома. В первом случае наблюдается окклюзия отверстий Люшка и Мажанди, что сопровождается серьезными нарушениями ликвородинамики. Второй вариант не имеет таких патологий, поэтому протекает в более благоприятной форме. В нейрохирургии важное значение имеет анатомическая классификация порока, согласно которой выделяют 2 формы синдрома Денди-Уокера:
- Полная. Характеризуется отсутствием червя мозжечка, наличием связи между полостью четвертого желудочка и мозжечково-мозговой цистерной.
- Неполная. Проявляется частичным недоразвитием мозжечкового червя в его нижней части, за счет чего коммуникация с большой цистерной реализуется не на всем протяжении.
Симптомы
Основное проявление синдрома Денди-Уокера – гидроцефалия, возникающая в первые месяцы жизни ребенка. Степень выраженности симптоматики зависит от варианта синдрома, скорости прогрессирования нарушений, общего состояния новорожденного. К ранним признакам относят беспокойное поведение, монотонный крик, нарушение питания и частые срыгивания. Внешне обнаруживается выбухание родничков, расширение черепных швов, быстрое увеличение окружности головы.
Характерный признак гидроцефалии при синдроме Денди-Уокера – симптом заходящего солнца: при взгляде вниз радужка частично скрывается нижним веком, над ней появляется белая полоска склеры. Неврологические симптомы также представлены нистагмом, экзофтальмом, косоглазием. Нередко у новорожденных отмечается судорожный синдром, парезы и параличи конечностей. При прогрессировании болезни характерно отставание в психомоторном развитии.
Осложнения
В 65-68% случаев синдром Денди-Уокера сопровождается другими структурными неврологическими аномалиями. Чаще всего диагностируется агенезия мозолистого тела, стеноз Сильвиева водопровода, гетеротопия извилин коры мозжечка. При тяжелых формах заболевания определяется недоразвитие ствола головного мозга, что сопряжено с глубоким угнетением витальных функций и высоким риском младенческой летальности.
До 55% детей имеют сопутствующие врожденные синдромы: Уокера-Варбурга, PHACE, Ritscher-Schinzel. Характерны различные формы хромосомных аберраций: делеции, дупликации, трисомии и триплоидии. Поражение кардиоваскулярной системы проявляется септальными дефектами и нарушениями постнатальной гемодинамики. Реже определяются офтальмологические пороки: ретинальная дисплазия, микрофтальмия.
Диагностика
Первичное обследование ребенка с неврологическими нарушениями проводится врачом-педиатром или неонатологом. При гидроцефалии и подозрении на синдром Денди-Уокера к диагностике подключается детский невролог, нейрохирург. Физикальный осмотр выявляет неспецифические признаки внутричерепной гипертензии, нарушения моторного развития, сопутствующие аномалии. Для постановки окончательного диагноза проводится:
- Нейросонография. При ультразвуковом сканировании головного мозга определяется крупное кистозное образование в задней части черепной коробки. На УЗИ мозжечковый червь не визуализируется, недоразвитые полушария мозжечка раздвинуты, желудочковая система мозга расширена и деформирована.
- МРТ головного мозга. Магнитно-резонансная томография в сагиттальной и аксиллярной проекции демонстрирует расширение четвертого желудочка, грубые нарушения развития мозжечка, другие структурные аномалии.
- Эхокардиография. УЗИ сердца рекомендовано всем детям с синдромом Денди-Уокера, поскольку он нередко ассоциирован с врожденной кардиальной патологией. По результатам ЭхоКГ можно определить аномалии развития клапанного аппарата, дефекты внутрисердечной перегородки, патологии расположения крупных сосудов.
- Кариотипирование. При комбинированных врожденных пороках развития необходима консультация генетика и тщательное изучение генетического материала ребенка. Углубленное исследование направлено на диагностику хромосомных аберраций.
- Пренатальная диагностика. При проведении УЗИ плода удается предположить аномалию на сроке 15-16 недель, более четкая визуализация структур IV желудочка возможна после 22 недели гестации. При сочетании патологии с расщелинами лица эхосонография может быть недостаточно информативна.
Дифференциальная диагностика
При постановке диагноза необходимо дифференцировать синдром с гипоплазией мозжечка другой этиологии, ретроцеребральными кистами и расширениями большой мозговой цистерны. Патогномоничным признаком синдрома Денди-Уокера считается дефект червя мозжечка, который не возникает при других вариантах пороков ЦНС. Проводится дифференциальная диагностика с арахноидальными кистами, которая требует дополнительных инструментальных методов исследования.
Лечение синдрома Денди-Уокера
Консервативная терапия
Наибольшую опасность для жизни и здоровья младенца представляет гидроцефалия, для коррекции которой показано лечение в отделении интенсивной терапии. Назначается массивная дегидратационная терапия, которая включает разные типы диуретиков, гипертонические инфузионные растворы. Усилия врачей направлены на уменьшение ликворопродукции и нормализацию внутричерепного давления, чтобы предупредить отек мозга и компрессию церебральных структур.
Хирургическое лечение
При неэффективности консервативных методов и неуклонном прогрессировании гидроцефалии требуется помощь детских нейрохирургов. Основным методом коррекции при синдроме Денди-Уокера являются шунтирующие операции с имплантацией дренажных трубок или клапанных регулируемых систем. Существует несколько типов шунтирования: вентрикуло-перитонеальное, вентрикуло-атриальное, вентрикулоцистерностомия.
Для коррекции окклюзии ликворных путей применяются методы эндоскопической вентрикулостомии, которые восстанавливают нормальный отток цереброспинальной жидкости, устраняют острую неврологическую симптоматику. При гипертензионно-гидроцефальном кризе показана вентрикулярная разгрузочная пункция, которая относится к вариантам экстренного временного дренирования.
Прогноз и профилактика
Исход заболевания зависит от глубины поражения ЦНС, скорости нарастания гидроцефалии, наличия сочетанной патологии. Нарастающая гипертензия и грубые неврологические пороки в 90% случаев становятся причиной смерти пациента в первые годы жизни, для выживших детей с неполным вариантом аномалии прогноз более благоприятный. Вследствие недостаточной изученности этиопатогенеза болезни эффективные меры профилактика пока не разработаны.
1. Случая мальформации Денди-Уокера с доношенной беременностью и родами / Е.В. Беляева, Л.В. Лапшина, Е.В. Шапошникова, А.А. Молгачев // Журнал неврологии и психиатрии. – 2018. – №2.
2. Аномалия Денди-Уокера – редкая причина сирингомиелии у взрослых /Г.Ю. Евзиков// Неврология, нейропсихиатрия, психосоматика. – 2017. – №9.
3. Синдром Денди-Уокера у новорожденных/ Л.А. Петрова, А.В. Розанов, Ю.И. Барашнев, В.О. Панов// Российский вестник перинатологии и педиатрии. – 2010. – №1.

Синдром Денди – Уокера — врожденная аномалия развития, характеризующаяся нарушениями строения мозжечка и путей оттока ликвора. Первые признаки болезни выявляются при проведении планового ультразвукового исследования в период беременности. У детей после рождения нарушена моторика, присутствуют явления гидроцефалии, а также другие симптомы. Для устранения признаков расстройства назначаются медикаменты, снижающие тонус мышц и уменьшающие внутричерепное давление. Возможно проведение хирургических операций для улучшения оттока спинномозговой жидкости.
Причины развития
Выявить причину развития заболевания не всегда удается. Специалисты отмечают ряд признаков, приводящих к симптомам болезни:
- увеличение объема четвертого желудочка, что приводит к скоплению большого объема ликвора;
- нарушения оттока спинномозговой жидкости в результате зарастания или полного отсутствия путей для этого;
- объемные опухолевые образования в задней черепной ямке;
- нарушение строения мозжечка. У детей выявляется увеличение объема полушарий на фоне атрофии червя.
Указанные изменения в строении структур головного мозга связаны с генетическими аномалиями у плода. Они возникают на фоне перенесенных внутриутробных инфекций (краснуха, цитомегаловирусная патология, герпес), аутоиммунных или обменных нарушений у беременной. При длительной интоксикации (табакокурение, алкоголизм) в период беременности риск возникновения синдрома Денди – Уокера также увеличивается.
Клинические проявления
Первые признаки болезни могут быть выявлены на УЗИ во время прохождения беременной женщиной ультразвукового скрининга. При этом специалист определяет следующие патологические изменения:
- уменьшение размера мозжечка, в первую очередь, червя, расположенного между его полушариями;
- кистозное образование в задней черепной ямке;
- увеличение размера четвертого желудочка.
Эти признаки могут наблюдаться при других наследственных заболеваниях — синдроме Марфана, Дауна и пр. Дифференциальная диагностика в период внутриутробного развития ограничена.
После рождения симптоматика синдрома Денди – Уокера представлена следующим образом:
- диспропорции головы ребенка. Она имеет увеличенный размер, что связано с гидроцефалией;
- краниотабес, характеризующийся размягчением костей в области родничков;
- увеличение размеров родничков;
- гидроцефалия, приводящая к утренним головным болям, тошноте, рвоте и слабому крику ребенка;
- нарушения формирования челюсти и носовых раковин;
- нистагм, проявляющийся непроизвольными колебательными движениями глазных яблок;
- нарушения согласованных движений рук и ног, а также мелкой моторики;
- общее беспокойство.
Дети в возрасте более 1 года характеризуются тревожностью и раздражительностью. После пробуждения они жалуются на головную боль. У них появляются тошнота, рвота и моторные нарушения. В процессе взросления формируются умственная отсталость и задержка психомоторного развития. Патология часто сопровождается аномалиями развития внутренних органов: сердца, легких и др.
Негативные последствия
Осложнения заболевания развиваются на фоне основных симптомов патологии. К негативным последствиям болезни относятся:
- задержка психомоторного развития разной степени выраженности;
- умственная отсталость;
- очаговая неврологическая симптоматика: гипертонус мышц рук и ног, гиперкинез отдельных мышечных групп, нарушенная координация движений;
- хроническая сердечная недостаточность;
- заболевания мочевыделительной системы и др.

Синдром Денди – Уокера приводит к инвалидности с рождения. 40–50% пациентов не доживает до 6 месяцев. Аналогичный прогноз наблюдается при других наследственных заболеваниях — синдроме Ретта и пр.
Диагностические мероприятия
Основная задача врача — проведение пренатальной диагностики патологии. Ультразвуковое исследование плода позволяет заподозрить болезнь и провести дифференциальную диагностику с другими врожденными аномалиями (синдром Эдвардса и др.). Подтвержденный диагноз является показанием для прерывания беременности при согласии женщины. После рождения ребенка диагностические мероприятия проводятся по определенному алгоритму:
- Сбор имеющихся жалоб и внешний осмотр ребенка. Специалист выявляет аномалии строения костей черепа, увеличение размера родничков и головы, а также неврологическую симптоматику. Последняя представлена гипертонусом мышц и нарушениями мелкой моторики.
- При беседе с родителями, выявляются факторы риска развития патологии — перенесенные в период беременности инфекционные заболевания, алкоголизм, наркомания и др.
- Проводится УЗИ структур центральной нервной системы и внутренних органов. Обследование позволяет выявить аномалии развития и определить тактику их лечения.
- Компьютерная или магнитно-резонансная томография используются для исследования состояния внутренних органов. Методы заменяют УЗИ, так как обладают большей диагностической ценностью для врачей.
- Консультации с педиатром, неврологом, кардиологом, офтальмологом и другими специалистами при наличии показаний.
Интерпретировать результаты проведенных исследований должен только врач. Неправильная постановка диагноза — причина прогрессирования нарушений работы головного мозга и внутренних органов, приводящих к тяжелым осложнениям.
Подходы к лечению
Полное выздоровление невозможно. Терапия позволяет увеличить продолжительность жизни и предупредить развитие осложнений заболевания. Она включает в себя консервативные методы и хирургические вмешательства.
Операции направлены на восстановление оттока ликвора. С этой целью врачи проводят шунтирование — создают дополнительные пути для спинномозговой жидкости. Вмешательство на четвертом желудочке головного мозга также уменьшает признаки гидроцефалии. Кроме этого, операции проводятся при пороках сердца, почек и других внутренних органов.
Лечение медикаментами устраняет основные проявления болезни. Общий прогноз не меняется. При выраженном мышечном гипертонусе назначают препараты для его уменьшения. Кроме того, врач может выписать другие медикаменты:
- кортикостероидные гормоны — уменьшают количество образующейся спинномозговой жидкости;
- препараты, расширяющие кровеносные сосуды, снижают внутричерепное давление за счет улучшения оттока ликвора;
- диуретические средства;
- бета-блокаторы уменьшают отечность нервной ткани и внутричерепное давление.
К средствам поддерживающей терапии относят:
- обезболивающие препараты из группы нестероидных средств, позволяющих снизить выраженность боли на фоне внутричерепной гипертензии;
- аминокислотные средства, улучшающие обменные процессы в нервных клетках и защищающие их от повреждений;
- успокоительные препараты, используемые при повышенной возбудимости и гиперактивности ребенка.
Если у больного наблюдаются сопутствующие поражения внутренних органов, то врач подбирает средства для уменьшения их симптоматики.
Важно помнить, что все препараты имеют определенные показания и противопоказания, которые необходимо учитывать при их назначении. Препараты назначает только врач!
В терапии используют немедикаментозные подходы: лечебный массаж и физкультуру, а также водные процедуры. Указанные методы нормализуют мышечный тонус, снимают спазм мускулатуры и уменьшают выраженность болевого синдрома.
Как предупредить заболевание?

Специфическая профилактика наследственных болезней отсутствует. Врачи рекомендуют беременным женщинам соблюдать следующие принципы в период вынашивания плода:
- избегать воздействие токсических веществ на работе, а также отказаться от курения, употребления спиртных напитков и наркомании;
- своевременно обращаться в женскую консультацию для наблюдения у гинеколога;
- соблюдать профилактику инфекционных заболеваний. Если женщина имеет хроническую цитомегаловирусную или герпетическую инфекцию, в период беременности следует поддерживать высокую активность собственного иммунитета;
- не пропускать ультразвуковой скрининг и другие назначения врача.
Простые советы помогают снизить риск развития наследственных патологий, в том числе синдрома Клайнфельтера и др. При наличии в семье генетических болезней в период планирования беременности рекомендуется проконсультироваться с врачом-генетиком. Он проведет обследование женщины и мужчины, составив список рекомендаций, снижающих вероятность рождения ребенка с аномалиями развития.
Специфическая профилактика отсутствует. Основная задача родителей — пройти подготовку к беременности и устранить факторы риска.
Синдром Денди – Уокера — тяжелое наследственное заболевание с неблагоприятным прогнозом. Около 50% новорожденных не доживает до полугода и умирает на фоне недостаточности внутренних органов или тяжелого поражения головного мозга.
Своевременное внутриутробное выявление болезни с помощью УЗИ-скриннига позволяет провести медицинское прерывание беременности и предупредить рождение больного ребенка. Эффективная терапия болезни невозможна и носит симптоматический, поддерживающий характер.
Аномалии формирования центральной нервной системы — отнюдь не редкость. Эта патология занимает второе место по частоте после врожденных дефектов развития сердца и сосудов и в большинстве случаев представлена гидроцефалией самого разного происхождения. Помимо тяжелых нарушений, сопровождающих патологическое развития мозга, пороки ЦНС несут высокий риск смертельного исхода, а по данным статистики они лидируют по числу смертей в младенческом возрасте.
Синдром Денди-Уокера — одна из разновидностей нарушения формирования головного мозга еще во время внутриутробного развития. И хотя частота его относительно невелика (всего 1 случай на 25-30 тысяч младенцев), диагностируется порок едва ли не у каждого десятого малыша с врожденной гидроцефалией, которая и служит одним из основных проявлений патологии.
Синдром Денди-Уокера — это порок задней черепной ямки, при этом основные структурные изменения касаются мозжечка, ликвороотводящих путей, четвертого желудочка мозга. Аномалия диагностируется во время беременности посредством ультразвукового осмотра, после чего женщине может быть предложено прерывание беременности по медицинским показаниям.

Конечно, любые отклонения в развитии плода — это всегда большой стресс и переживания для родителей, однако в случае врожденного порока мозга надеяться на чудо не приходится — прогноз серьезный, а смертность высока. Малыши с сочетанными пороками мозга и других органов погибают в раннем возрасте как от мозговой дисфункции, так и от присоединившейся инфекции.
Аномалия Денди-Уокера часто сочетается с другими нарушениями и генетическими заболеваниями, зачастую несовместимыми с жизнью — микроцефалия (недоразвитие полушарий мозга), мозговые грыжи. У младенца может быть диагностирован генетически обусловленный поликистоз почек, недоразвитие зрительных нервов и глазных яблок со слепотой, аномалии сердечно-сосудистой системы.
Все эти неблагоприятные факторы, возможность сочетанной патологии многих органов делают синдром Денди-Уокера серьезнейшей проблемой в случае, если малышу дадут возможность родиться. Лечение патологии, как правило, симптоматическое, направленное на поддержание главных систем жизнеобеспечения и борьбу с инфекционными осложнениями. В редких случаях применяют хирургическую операцию, которая лишь облегчает явления гидроцефалии, но не ликвидирует ее полностью.
Почему возникает синдром Денди-Уокера?
Причины аномалий развития задней черепной ямки до сих пор не выяснены, однако выделен ряд факторов, способствующих подобным врожденным порокам:
- Инфицирование во время беременности цитомегаловирусом, перенесенная краснуха;
- Употребление алкоголя, курение, наркомания во время беременности;
- Экстрагенитальная патология, особенно — сахарный диабет у будущей мамы.
Под действием перечисленных причин или среди полного благополучия может возникнуть спонтанная мутация в генах, предрасполагающая к нарушению развития мозга. Особенно высок риск пороков при действии неблагоприятных факторов во время первого триместра гестации, когда и происходит закладка основных структур центральной нервной системы.
В части случаев синдром Денди-Уокера носит наследственно-обусловленный характер, то есть возникает из-за дефекта генов и может передаваться по наследству как рецессивный признак. Если первая беременность протекала с формированием этой патологии, то риск повторного порока в последующем возрастает до 25%.
Что происходит с мозгом при синдроме Денди-Уокера?
Анатомически классический вариант мальформации задней черепной ямки включает:
- Гидроцефальный синдром разной степени выраженности;
- Кистозную полость в задней части черепа с расширенным четвертым желудочком мозга;
- Отсутствие или недоразвитие червя мозжечка, недоразвитие его полушарий.
Червь мозжечка — это структура, расположенная между его половинами и несущая в себе проводящие нервные волокна. При аномалии Денди-Уокера он может быть представлен небольшой щелью или широким пространством между гемисферами органа. При неполном отсутствии червя щелевидное расширение образуется лишь в нижней его части. На фоне патологии этого отдела наблюдается недостаточное развитие и мозжечковых полушарий.
Именно дефект мозжечкового червя в виде расщелины считается характерным признаком аномалии Денди-Уокера, позволяющим отличать ее от недоразвития на фоне других пороков мозга.

Киста четвертого мозгового желудочка может самопроизвольно вскрыться в 3-ий желудочек или субарахноидальное пространство. В этом случае симптомы окклюзии ликворных путей несколько ослабнут. Выраженность гидроцефального синдрома вариабельна — от небольшого расширения желудочковой системы до высокой степени окклюзионной гидроцефалии с отсутствием возможности для циркуляции ликвора.
Многие специалисты отмечают, что у большинства малышей с синдромом Денди-Уокера при рождении гидроцефалии как таковой нет, а формируется она и прогрессивно нарастает в течение первых нескольких месяцев жизни, поэтому факт отсутствия гидроцефального синдрома сразу после родов при наличии диагностированной внутриутробно патологии не является поводом для пересмотра диагноза и необоснованных надежд, с этим связанных.
Более, чем в половине случаев синдрома Денди-Уокера у детей помимо описанных структурных аномалий обнаруживаются и другие дефекты мозга — недоразвитие или отсутствие мозолистого тела, мозговые кисты, недоразвитие или отсутствие извилин, смещения серого вещества относительно его правильной локализации, что еще больше усугубляет течение и без того тяжелой патологии.
По данным МРТ было выделено несколько разновидностей синдрома Денди-Уокера:
- Классический тип аномалии — задняя черепная ямка расширена, четвертый желудочек кистозно изменен, червь мозжечка частично или полностью недоразвит, полушария его гипоплазированы, а намет находится выше, чем в норме, желудочковая система не сообщается с подпаутинным пространством, часто наблюдаются мозговые кисты и отсутствие мозолистого тела, практически у всех пациентов есть гидроцефалия, возможно сдавление стволовых структур. Порок проявляется клинически уже с рождения и имеет неблагоприятный прогноз.
- Вариант Денди-Уокера — морфологические признаки выражены меньше, чем при классической форме, гипоплазирован нижний отдел червя мозжечка, желудочки сообщаются с кистой и ликворными пространствами, обеспечивая отток ликвора, поэтому гидроцефалия наблюдается редко. Задняя черепная ямка имеет нормальные размеры, стволовые структуры не сдавливаются.
- Киста кармана Блейка — расширение желудочковой системы с гидроцефальным синдромом, киста расположена под или за мозжечком, червь развит относительно хорошо. Четвертый желудочек расширен, но не сообщается с затылочной ликворной цистерной.
- Mega cisterna magna — вариант очагового расширения подпаутинного пространства в задней и нижней частях задней ямки черепа с увеличением объема затылочной цистерны, которая сообщается с четвертым желудочком и субарахноидальным пространством.
Проявления заболевания
Симптоматика синдрома Денди-Уокера разнообразна. Возможны как практически нормальное развитие ребенка после рождения, так и грубые неврологические изменения, влекущие тяжелую инвалидность и даже смерть. По некоторым данным, нормальное развитие интеллекта бывает в половине случаев изолированного порока, возможно даже случайное обнаружение синдрома при обследовании взрослых.

дети с синдромом Денди-Уокера
Внутриутробное течение патологии определяется степенью поражения мозга, нарастанием гидроцефалии, наличием других пороков развития. Прогноз значительно хуже при диагностике синдрома до рождения. При глубоких нарушениях в формировании мозга на первый план среди других проявлений выступает гидроцефальный синдром:
- Увеличение диаметра головы;
- Выбухание родничка.
Увеличение диаметра черепа происходит, главным образом, за счет затылочной области, в которой образуется киста, вызывающая истончение и растяжение костной основы. При выраженной гидроцефалии голова малыша активно растет на протяжении первых двух месяцев, параллельно происходит расхождение швов между костями спереди или в заднем отделе. Кроме того, характерны:
- Повышение нервной возбудимости (рефлексов);
- Глазодвигательные расстройства — нистагм, косоглазие;
- Приступы остановки дыхания;
- Парез лицевого нерва.
Симптомы мозжечковых нарушений у новорожденных выявить невозможно, и даже тяжелый дефект формирования мозжечковых структур далеко не всегда вызывает значимые признаки атаксии (двигательных расстройств), которая регистрируется всего у трети пациентов.
Дети с гидроцефалией в раннем возрасте беспокойны, плохо спят, характерен монотонный крик, усиление рефлексов, плавающие движения глаз и их закатывание, выраженность сосудов роговицы, заметная подкожная венозная сеть по мере роста размеров головки. Спонтанная двигательная активность новорожденных может быть ослаблена, возможны судороги и тетрапарез из-за гипертонуса мышц.
В более старшем возрасте становится заметным отставание в психическом и интеллектуальном развитии, дети не могут обучаться, быстро устают, плохо усваивают новую информацию, что делает процесс адаптации крайне затруднительным. В тяжелых случаях обучение невозможно совсем, в связи с чем ребенок нуждается в постоянной посторонней помощи, уходе и рассмотрении вопроса об инвалидности.
Моторное развитие заметно замедлено. При тяжелых формах аномалии дети не могут своевременно научиться переворачиваться, ползать, садиться и ходить, не удерживают взгляд на игрушках, быстро устают и часто плачут. Возможны нарушения питания с гипотрофией, общее снижение иммунитета, частые инфекционные заболевания.
Сочетание порока нервной системы с другими аномалиями развития органов предрасполагает к серьезным осложнениям, в числе которых не только мозговая дисфункция, слабоумие, судорожный синдром, но и сердечная недостаточность, склонность к пневмониям при пороках сердца, хроническая почечная недостаточность и уремия при врожденном поликистозе, что усугубляет явления отека мозга и может послужить причиной гибели пациента.
При тяжелой окклюзионной гидроцефалии смерть может наступить в раннем младенчестве от отека головного мозга, фатальных аритмий, остановки дыхания на фоне компрессии стволовых структур, тяжелой пневмонии и других инфекционных осложнений.
У взрослых возможно постепенное нарастание гидроцефалии с краниалгиями, снижением памяти и внимания, раздражительностью, склонностью к депрессии, утренней тошнотой и рвотой на высоте головной боли. В тяжелых случаях бывает судорожный синдром. Возможны проблемы с координацией и выполнением мелких движений, неуверенность при ходьбе, зрительные расстройства.
Диагностика и лечение
Диагностика синдрома Денди-Уокера основывается на результатах ультразвукового осмотра, при этом важно обнаружить аномалию еще во время эмбрионального развития. УЗИ становится информативным после 18 недели гестации, но в некоторых случаях заподозрить патологию можно и раньше — уже на 14-15 неделях эмбрионального развития.

Диагностическими критериями при аномалии задней черепной ямки считаются:
- Наличие крупной кистозной полости, включающей четвертый мозговой желудочек, в задней части черепа;
- Отсутствие или аномальное развитие червя мозжечка;
- Гипоплазия мозжечковых гемисфер, наличие широкой щели между ними;
- Расширение желудочковой системы (гидроцефалия).
Для постановки диагноза синдрома Денди-Уокера необходимы:
- УЗИ (нейросонография);
- МРТ для определения анатомических особенностей четвертого желудочка мозга;
- Консультация офтальмолога;
- Осмотр нейрохирурга;
- УЗИ сердца для исключения врожденных аномалий;
- Консультация генетика и определение кариотипа при возможных генетических мутациях.
Лечение патологии определяется симптоматикой и тяжестью проявлений. Если гидроцефалии нет, а внутричерепное давление в пределах нормы, то оправдано динамическое наблюдение невролога, педиатра или нейрохирурга, каких-либо медикаментов не требуется.

шунтирование для нивелирования гидроцефалии
При нарастании гидроцефалии и внутричерепного давления показаны шунтирующие хирургические операции для отвода ликвора из черепа в грудную или брюшную полость. Медикаментозное лечение включает применение диуретиков (диакарб, маннитол), ноотропных средств (пирацетам, пантогам), антиконвульсантов (депакин).
В случае гипертонуса показаны физиотерапевтические и водные процедуры, массаж, специальные упражнения. Важен тщательный уход и постоянное наблюдение за малышом, создание спокойной обстановки при беспокойном поведении и нарушениях сна.
При тяжелых формах течения патологии с отставанием в интеллектуальном развитии детям показана работа с дефектологами-педагогами, психологом по индивидуальной программе, исключающей избыток информации и умственное перенапряжение.
Прогноз при синдроме Денди-Уокера зависит от ряда причин: времени установления диагноза, наличия других пороков и хромосомных болезней, степени окклюзии ликворных путей. Смертность и заболеваемость после рождения выше в тех случаях, когда аномалия сочетается с другими дефектами и обнаружена до рождения малыша.
Гидроцефалия и внутричерепная гипертензия — ключевые моменты в определении прогноза, которые влияют и на развитие пациента, и на продолжительность и качество его жизни. В случае изолированного поражения мозга без признаков гидроцефалии прогноз благоприятный. Ребенок может развиваться по возрасту, а иногда аномалия и вовсе выявляется у взрослых при обследовании по поводу других причин.
В связи с тем, что причины порока так и не выяснены, проводить специфическую профилактику не представляется возможным. Конечно, нужно соблюдать здоровый образ жизни, особенно, женщинам, планирующим беременность или уже забеременевшим с исключением вредных привычек, неблагоприятных влияний внешней среды. Важно своевременно выявить и пролечить цитомегаловирусную инфекцию, герпес, а в случае краснухи, которой женщина заболела при беременности, врачи предложат аборт по медицинским показаниям из-за высокого риска сочетанных пороков.
Решать вопрос о сохранении беременности в том случае, если синдром возник случайно, у плода абсолютно здоровой женщины, придется будущей маме и ее семье. Решение всегда дается сложно, но следует знать, что аномалия нервной системы и нормальное развитие и рост ребенка — скорее, исключение из правил.
В абсолютном большинстве случаев детям и родителям приходится бороться с гидроцефалией, зачастую требуется не одна дорогостоящая и сложная операция, тогда как ее эффективность и прогноз все равно могут оставаться сомнительными.
Видео: примеры детей с синдромом Денди-Уокера
Читайте также: